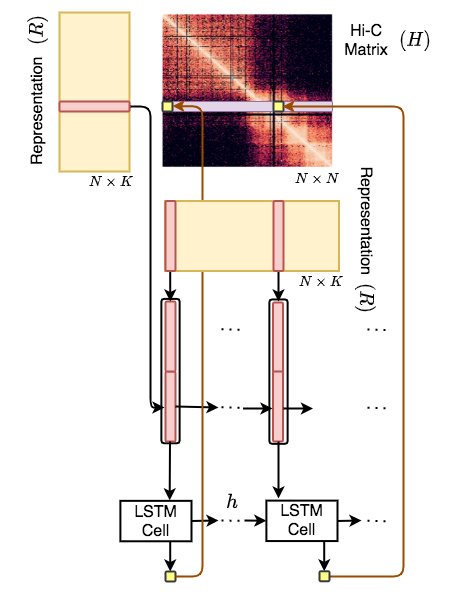
Hi-C-LSTM is a framework to build Hi-C intrachromosomal representations which are useful for element identification and in-silico alterations.
![]()
![]()
This might not be SOTA for Hi-C representation learning anymore. Refer to Akita, Orca, and Origami. Researchers interested in a preliminary hierachical version of Hi-C-LSTM can check HirarChy and those looking to apply contastive learning to Hi-C data can get started with HiCClip.
The following software was installed on Ubuntu 16.04
- Python 3.7.10
- CUDA 10.1 with libcudnn.so.7
- torch 1.8.0
- captum 0.3.1
- numpy 1.21.0
- pandas 1.2.4
- scipy 1.7.0
- matplotlib 3.4.2
- tensorboard 2.5.0
- seaborn 0.11.1
Install the above dependencies using installers like pip. The typical install time is about 1 hour. No non-standard hardware is required.
- get HiC data: Download Juicer Tools and run ./extract_chromosomes.sh. Specify right path for juicer tools jar.
- run compute_genome_length.py to create file with rounded, cumulative chromosome lengths
- Use the
hic_chr22.txtfile as input for demo - Partition the file based on training and testing needs
- Model parameters, hyperarameters, and output directories can be changed in
./code/config.py.
./code/train_model.py
- Specify the
model_nameof your choice - In the
DataLoader, under theget_data_loader(cfg, cell)function, specify the chromosomes to be used - For the demo case, use chromosome 22. Change the directory of input Hi-C data to
.data2/ - Expected output is a trained model called
model_name. Expected training time is less than 8 minutes per epoch on GeForce GTX 1080 Ti GPU.
./code/test_model.py
- Use the trained model
model_nameto test on the remainder of the chromosome 22 data - Expect the MSE of the model as the output along with predictions and representations. Expected testing time is less than 5 minutes.
- Extract the representations of size
representation_sizefrom the prediction file - Align them with the genome at 10Kbp resolution
- Use for downstream tasks of preference like classification of genomic phenomena and in-silico mutagenesis.