This is a brief introduction of RPA application and the solution of several common problems; Also, I will list important input keywords and help you with a better understanding of the results of the simulations.
The memory cost of RPA is extremely huge, therefore, such kind of "stuck" would often happen
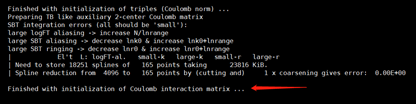
get stuck at "Finished with initialization of Coulomb interaction matrix" and
get stuck at RPA correlation energy calculation over all k-points. If your output file ends at the above position, and you also receive some error message like “MPI error”, it’s always the consequence of insufficient memory. There are two memory “peaks” during the entire calculation, as you can see above, the initialization of Coulomb matrix(based on auxiliary basis sets, which are always about 5 to 7 times compared to original numerical atomic orbitals) and the summation of RPA correlation energy over all k-points. The coulomb matrix V (also the response function ) has such dimension
, which means the growth of basis sets (more atoms, larger basis sets) causes much more computational time rather than the growth of k-points. Right now, NAO-VCC-3Z basis might be the largest basis sets you may want to use; NAO-VCC-4Z sometimes works but only for lighter elements ( <10 ) and less atoms in unit cell (up to 4-6 atoms).
You can just increase the nodes and cores directly, however, when it comes to large systems with more atoms, heavier elements or larger basis sets (tight, NAO-VCC-4Z) which might demand hundreds even thousand of CPUs, It’s not a wise choice wasting your time waiting for available resources. I suggest multi-thread calculation in FHI-AIMs, and you have to set this variable in your script for submitting:
export OMP_NUM_THREADS = n
where n is the number of threads you want to use; Generally the default value of n equals to 1 for efficiency, but right now you can adjust this variable to finish your calculation with less nodes, but more real time.
There is another possibility of this error, rare but sometimes happen: In some computer clusters, the manager would make full use of the nodes and allow different tasks on the same node; This might cause the memory contention problem, which also lead to insufficient memory error; If so, make sure you own task has occupied the entire node.
This section would give out the meaning of important output energy. The RPA total energy (for example RPA@PBE) is defined as
All of these components could be found in output file, in the following output block:
--------------------------------------------
Periodic RPA total energy calculation starts ...
-----------------------------------------------------------------------
Start to calculate the periodic RPA correlation energy ...
First valence state in the frozen-core algorithm : 11
| Main-group Transition-metal
| Atom number :: 4 2
| n_frozen_shell :: 2 2
The first valence state in the frozen-core algorithm : 11
using n_freqs_block= 100 , n_blocks= 1
1 1 0.00000000 0.00000000 0.00000000 -7.15924286
2 2 0.00000000 0.00000000 0.33333333 -8.52616004
4 3 0.00000000 0.50000000 0.00000000 -8.56225792
5 4 0.00000000 0.50000000 0.33333333 -8.46486161
7 5 0.50000000 0.00000000 0.00000000 -8.56225793
8 6 0.50000000 0.00000000 0.33333333 -8.46486161
10 7 0.50000000 0.50000000 0.00000000 -8.47976851
11 8 0.50000000 0.50000000 0.33333333 -8.42095460
-----------------------------------------------------------------------------
RPA correlation energy : -2.66630162 Ha, -72.55375855 eV
total_energy= -54801.20788844
en_xc= -3230.13247240
eex_energy= -3143.64802708
rpa_tot_en= -54787.27720168
--------------------------------------------------------------------
Exact exchange energy : -115.52694157 Ha, -3143.64802708 eV
DFT/HF total energy : -2013.90737353 Ha, -54801.20788844 eV
Exchange-only total energy : -2010.72912858 Ha, -54714.72344312 eV
RPA total energy : -2013.39543020 Ha, -54787.27720168 eV
RPA+SE total energy : -2014.07523038 Ha, -54805.77550573 eV
RPA+rSE total energy : -2013.64691788 Ha, -54794.12052960 eV
------------------------------------------------------------------------------
: DFT total energy from PBE functional, find
DFT/HF total energy or total_energy;
: exchange-correlation energy from PBE functional, find
en_xc;
: exact-exchange energy based on PBE functional, find
eex_energy;
: RPA correlation energy based on PBE functional, find
RPA correlation energy;
There are other information you might be interested in, such as number block in the middle
1 1 0.00000000 0.00000000 0.00000000 -7.15924286
2 2 0.00000000 0.00000000 0.33333333 -8.52616004
4 3 0.00000000 0.50000000 0.00000000 -8.56225792
......
This block presents the RPA correlation energy contribution of each irreducible k-points; Look at that from left to right, the first two columns are the index of k-points and irreducible k-points, and the following three columns are the coordinates of that k-points. One have to pay attention that these contributions should be combined with their weight factor to obtain the correct final RPA correlation energy.
Their are another two RPA energy with SE or rSE , where single excitation (SE) is involved to have a better description. The extra single excitation(SE) term would help correct RPA energy based on local or semi-local xc functionals, where RPA itself (usually PBE based) would systematically underbind the molecules and solids. Calculation of single excitation would not lead to extra computational cost. You can turn to this paper for more details: https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.153003
In general the RPA+SE or RPA+rSE is recommended to avoid underbinding situation.
This section is about the choice of basis sets. In FHI-AIMs, there are two basis sets widely used in RPA calculation: the traditional "Tier" basis, and "NAO-VCC-nZ" basis. These are both atomic-centered orbitals, while NAO-VCC-nZ is a special basis set that come from optimizing the RPA total energy of atom for each element (Right now only H - Ar available), and, can be applied a quite simple two-point extrapolation to complete basis set(CBS)
This is a sample of how to obtain complete basis set result CBS(3,4) via NAO3Z and NAO4Z. For more details, take a look at this paper from Igor Ying Zhang:
https://iopscience.iop.org/article/10.1088/1367-2630/15/12/123033/meta . I would suggest users perform RPA calculation under NAO-VCC-nZ basis sets if possible; Test the convergence of total energy or energy difference between phases carefully and choose NAO-VCC-2Z as first step to obtain an overall comprehension (physical property, phase stability, computational costs) of your studying object; Due to the absence of NAO-VCC-nZ basis beyond Ar, e.g. the transition metals, we can only use TIER basis at this moment, but you can still get reliable results using TIER basis.
The convergence of K-grids is much easier compared to basis sets. You may still find out that the convergence of total energy still require a large amount of k-points (to reach e.g. the common convergence criteria <1 meV/atom), while the energy difference between phases could dramatically converge with fewer k-points. If you are using NAO-VCC-nZ basis sets, you can also extrapolate results of two different k-grid to a infinite k-grid() called "CKM", also see https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.153003, but this is not necessary and results from direct calculation are always good enough to solve you problems.
RPA is a post-SCF calculation so you just need to add several extra tag to a traditional DFT input file control.in:
xc pbe
relativistic atomic_zora scalar #add this when element number > 18
k_grid <k_x> <k_y> <k_y>
total_energy_method rpa
frozen_core_postscf 2
#For accuracy / efficiency
prodbas_threshold 1.e-5
[attach the corresponding species defaults]
tag relativistic is always required when handling element that number > 18;
tag frozen_core_postscf is necessary, but the computational cost would increase when more occupied shells are considered;
tag prodbas_threshold is used to prevent the possible ill-conditioning of the auxiliary basis, the default value
Generally, two tags total_energy_method rpa and frozen_core_postscf n is enough for a standard RPA calculation, you can turn to FHI-AIMs manual for more details of these tags.
Because of the absence of force calculation in RPA (ongoing work), functions like geometry optimization and phonon calculation are not available right now. So, the most common use of RPA is benchmarking local and semi-local exchange-correlation functionals, and handling with puzzles originated from those polymorphs, like
Sixian Yang : smartyyy@mail.ustc.edu.cn