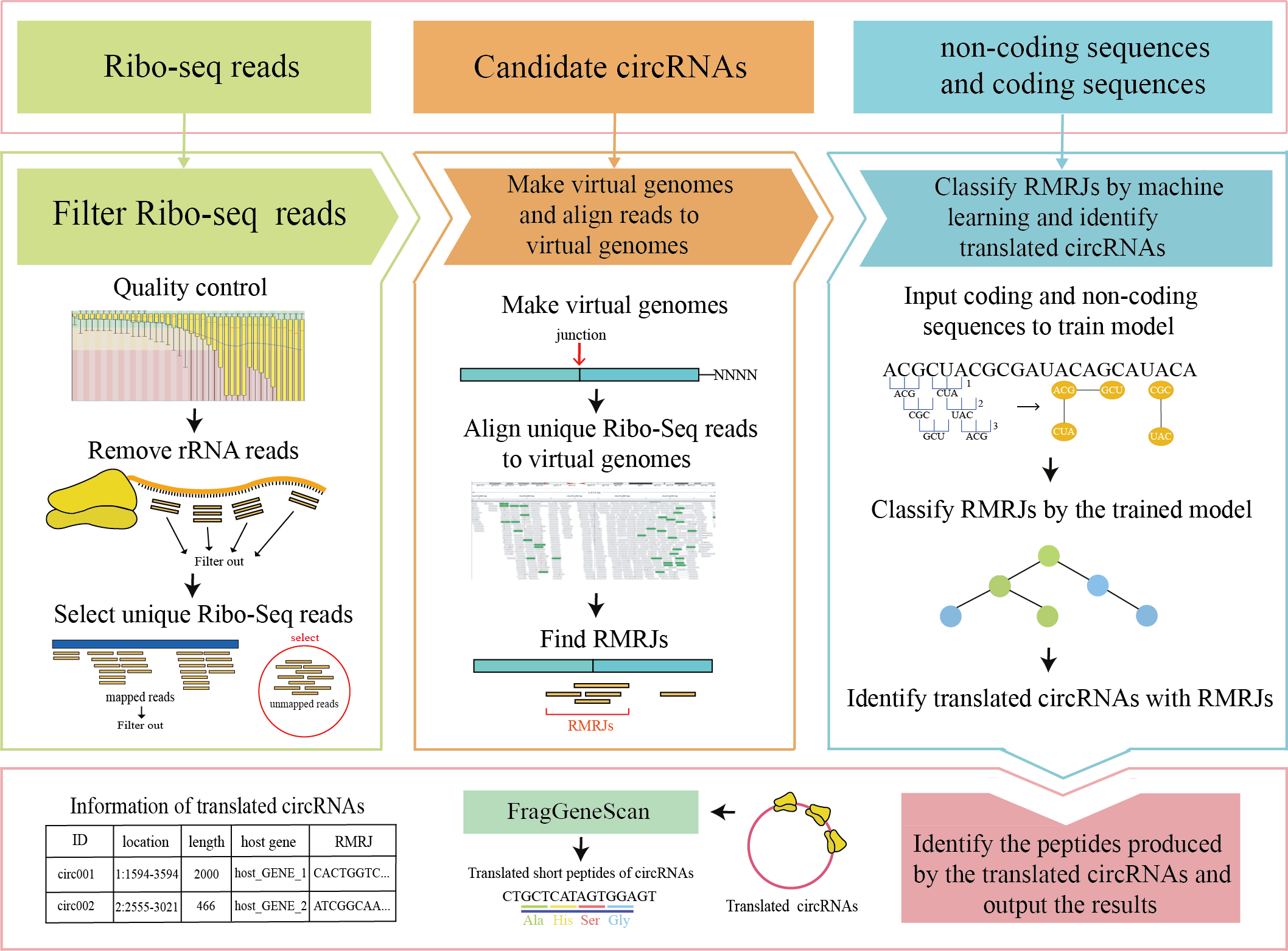
CircCode is a Python3-base pipeline for translated circular RNA identification. It automatically tandem links sequence in series and processes a given ribosome profile data (including quality control, filtering and alignment). Finally, based on J48 classification, the final translated circular RNA was predicted. The user only needs to fill in the given configuration file and run the python scripts to get the predicted translated circular RNA.
- Genome sequence (fasta format)
- Candidate circRNA sequence (fasta/bed format)
- rRNA sequence (fasta format)
- Adapter sequence (fasta format)
- Ribosome profiling data (sra format)
- Coding and non-coding sequence (fasta format)
- bedtools (v.2.26.0+): (https://bedtools.readthedocs.io/en/latest/)
- bowtie (v.1.2.2+): (http://bowtie-bio.sourceforge.net/index.shtml)
- STAR (v.2.7.1+): (https://github.com/alexdobin/STAR)
- Python3 (v.3.6.5+): (https://www.python.org/)
- R language (v.3.4.4+): (https://www.r-project.org/)
- Biopython (v.1.72+): (https://pypi.org/project/biopython/)
- Pandas (v.0.23.3+): (https://pypi.org/project/pandas/)
- BASiNET: (https://github.com/cran/BASiNET)
- Biostrings: (http://www.bioconductor.org/packages/release/bioc/html/Biostrings.html)

Open the terminal and input:
git clone https://github.com/PSSUN/CircCode.gitcd CircCode
./ Install.shNOTE: This step is optional, If you have already met all the required packages in your environment, you don't need to do this step, you can run the python script directly. You can also install the missing dependencies yourself. In the case that all dependencies are met, no compilation is required and all scripts can be run directly.
-
Fill the config file (https://github.com/Sunpeisen/CircCode/blob/master/config.yaml), input full path of each required file.
-
Run bash script on command line with your config file.
sh one_step_script.sh config.yaml-
Fill the config file (https://github.com/Sunpeisen/CircCode/blob/master/config.yaml), input full path of each required file.
-
Making virtual genomes
python3 make_virtual_genomes.py -y config.yamlThis script output two files: *.fa and *.gff, *.gff is the annotation information of *.fa. in tmp_file.
- Filter reads and compare to virtual genomes
python3 map_to_virtual_genomes.py -y config.yaml- Find RPF-covered region on junction (RCRJ) and classification of RCRJ by sequence features
python3 find_RCRJ_and_classify.py -y config.yamlYou can download the required sra file from NCBI-SRA, we also provide the other required files (includes genome.fa, genome.gtf etc.) in example.tar.xz. Fill in the path of the corresponding file into the project corresponding to config.yaml. Then follow the steps mentioned above to run each script.
When opening the config file in text format, there are some lines that need to be filled in, they are:
-
genome_name:
Each fasta file has its own name. Similarly, this value represents the name of the virtual genome generated by CircCode. You can fill in any value here in text form (we strongly recommend using only English letters). Note that you only need to fill in the name here, no suffix is required, for example, you should fill in 'textGenome' instead of 'textGenome.fa'.
-
genome_fasta:
You need to fill in the absolute path of the corresponding species genome here (not the relative path!)
-
genome_gtf:
You need to fill in the absolute path of the corresponding annotation file of species genome here (not the relative path!)
-
raw_reads:
CircCode's identification of circRNAs with translational potential relies on the support of Ribo-Seq data, and you need to fill in the absolute path of the Ribo-Seq data in sra format. The sra data can be your own sequencing data or downloaded from the NCBI public database. This supports inputting multiple sra data and making predictions at the same time. It is allowed if you only provide one sra file for prediction.
-
ribosome_fasta:
Here we need to provide rRNA data for the corresponding species in fasta format for filtering the Ribo-Seq data. Here you need to fill in the absolute path of this fasta file.
-
trimmomatic_jar:
The absolute path to the trimmomatic_jar file, we have provided the trimmomatic_jar file in CircCode, you just need to fill in the absolute path of this file on your computer.
-
circrnas:
The fasta file of the candidate circRNA, CircCode, is used to predict those circRNAs with translational potential from a given sequence of candidate circRNAs. This fasta file should contain all the candidate circRNAs and fill in the absolute path of this fasta file here.
-
riboseq_adapters:
The absolute path of the adapters file for Ribo-Seq data.
-
coding_seq:
The fasta file of the coding sequence in this species, if running on a small computer, in order to avoid memory overflow, this file should not be too large. Otherwise, you may get an error due to insufficient memory.
-
non_coding_seq:
The fasta file of the non-coding sequence in this species, if running on a small computer, in order to avoid memory overflow, this file should not be too large. Otherwise, you may get an error due to insufficient memory.
-
result_file_location:
Fill in the absolute path of a folder to hold the final run results.
-
tmp_file_location:
Fill in the absolute path of a folder to hold the temporary files.
-
reads_type:
The type of sequencing data, sequencing data is divided into single-ended and pairs-ended, corresponding, you need to fill in single or pair here
-
thread:
The number of threads running, only the number in the input int format is supported here, for example: 1 or 2 or 3 or 4 or 5...
NOTE:The test file is only used to test whether the software can run smoothly and does not represent the actual research results.
- 2019-10-14: Fixed a bug where the executable didn't run permission and caused an error.
- 2019-10-15: Update the Read.md
- 2019-12-16: Update the Read.md
- 2020-01-30: Remove the useless code of second script
- 2020-05-20: Update one_step_script.sh script
- 2020-07-06: Add a new parameter [merge] in yaml file
-
Q: In the prediction step, why does BASiNET not seem to have the expected results?
A: Make sure that the BASiNET package is properly installed on your computer. It is worth noting that BASiNET's dependency package rJava tends to have installation errors, ensuring that the dependencies are working fine.
-
Q: Do I need to fill out all the items in the yaml configuration file?
A: Yes, all projects need to be filled out. How to fill in the configuration file is explained in the previous section.
-
Q: I can't install R packages named 'rJava'.
A: Try apt-get install r-cran-rjava in ternimal.
-
Q: What result file means?
A: Please see here

- Zhang Jinwen’s ([email protected]) suggestion for the scripts was adopted.
- luhan125’s suggestion for the scripts was adopted.
If you encounter any problems while using CircCode, please send an email ([email protected] / [email protected]) or submit the issues on GitHub (https://github.com/Sunpeisen/circCode/issues) and we will resolve it as soon as possible.
