Peppy: Proteogenomic and Protein Identification Software A User’s Quick Start Guide
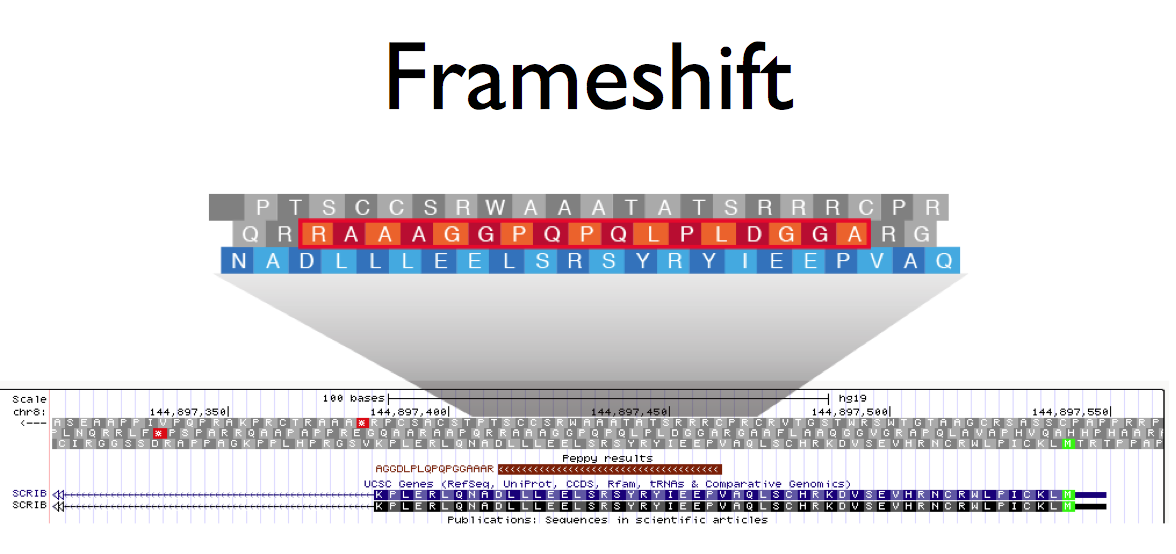 [above: example output of discovered of peptide from non-canonical gene expression]
[above: example output of discovered of peptide from non-canonical gene expression]
Welcome! You are probably using Peppy because you have a protein identification or proteogenomic project that you want to complete with good accuracy and you need it done fast. Peppy uses flexible core multi-threading, taking advantage of all of your computer’s processors, and advanced PSM scoring functions to help deliver high quality results in a reasonable amount of time. This Quick Start guide should help you get started using this basic features of the program.
- Your OS must have Java 1.6 or greater installed.
- Your memory allocation depends on the number of spectra and the database being searched. We’ve found allocating 8GB to be sufficient for many jobs.
- the Peppy.jar executable file
- a sample properties.txt file
- a file or a directory of files containing mass spectrometry data in DTA or PKL format
- a file or a directory of files containing DNA or protein sequences in FASTA format
- Create a folder named “Peppy”.
- Put the Peppy.jar executable file in the “Peppy” folder.
- In the “Peppy” folder, create a folder called “spectra”.
- Put a file or directory containing your mass spectrometry data in the “spectra” folder. Nested directories are acceptable.
- In the “Peppy” folder, create a folder called “sequences”.
- Put one or more files containing either DNA or protein sequences in the “sequences” folder.
- Put the properties file in the “Peppy” folder.
- If you wish, you can change the default properties in the properties.txt file (see 5. Properties, for a description of the default property settings).
- From the command line, open your computer’s terminal application and navigate to the “Peppy” folder. In the terminal window, run the following: Peppy Quick Start User’s Manual – Geneffects java -jar -Xmx12G Peppy.jar (note that “-Xmx12G” means that up to a maximum of 12 gigabytes of memory will be allocated to run Peppy)
- Peppy will run the data. You will get a message in the terminal window indicating the run has been completed (“done”).
- A new folder called “reports” will appear in the “Peppy” folder. In the “reports” folder, you’ll find a subfolder created for the run that was just performed, titled “spectra_uniquenumber” where uniquenumber is a large integer. In it, there will be three Peppy output files: • spectra_uniquenumber_properties.txt (a list of the properties used in that Peppy run) • spectra_uniquenumber_report.txt (a tab-delimited output file for that Peppy run) The text report can be viewed with a text editor or spreadsheet program (see 6. Peppy Report Column Definitions for a description of the report column headings).
The following are the default properties of the properties file we provide.
isSequenceFileDNA true
percursorTolerance 2.0
fragmentTolerance 0.3
For the purposes of getting you started, we included only these properties and default settings. Please note that many more properties can be customized using Peppy’s advanced features. Mass units for precursorTolerance and fragmentTolerance are in Daltons (Da).
spectrumID– a volatile tracking number (i.e. one that could change) assigned by Peppy and used for internal purposesfileLocus– for files that contain more than one spectrum, this is a zero-based index of the spectrum within the filespectrumMD5– an MD5 hash of spectrum file; this allows for spectrum tracking based on the spectrum’s content. Two spectra with the same content but different file names will still have the same MD5. Peppy Quick Start User’s Manual – GeneffectsFilePath– The system-specific file path ot the spectrum fileScore– the score for a peptide/spectrum match. This score is specific for the scoring algorithm.peptideMass– the calculated mass of the peptideprecursorNeutralMass– the mass of the precursor ion without the chargepeptideSequene– the amino acid sequence of matchpreviousAmionAcid– the n-terminal cleavage acid or “.”start– the start location for the matching peptide in that sequence filestop– the stop location for the matching peptide in that sequence fileSequenceName– the name of the sequence file; e.g. “chr4.fa”Strand– the strand (forward/reverse which corresponds to +/-) of the sequence file for the matching peptide (for DNA searches only)Rank Count– the number of matches at a particular rank for a spectrum. For example, say a spectrum has 3 top scoring peptides. Each peptide would have a rank of 1 and a rank count of 3.Ion Count– the number of theoretical peaks (b- and y-ions) produced by matching peptides that aligned with observed peaks in the MS/MS spectrumCharge– the charge of the spectrum
###Conclusion This Quick Start Guide is intended to get you familiar with Peppy’s main features. There are additional elements that are not described in this manual, including the ability to change many more properties, the ability to automatically run multiple jobs in sequence, the ability to score by other algorithms, and more. Peppy continues to be developed and refined so if you have suggestions for how it can be improved, please email Brian Risk at [email protected].