![]()
teemi: An open-source literate programming approach for iterative design-build-test-learn cycles in bioengineering



teemi, named after the Greek goddess of fairness, is a python package designed to make microbial strain construction reproducible and FAIR (Findable, Accessible, Interoperable, and Reusable). With teemi, you can simulate all steps of a strain construction cycle, from generating genetic parts to designing a combinatorial library and keeping track of samples through a commercial Benchling API and a low-level CSV file database. This tool can be used in literate programming to increase efficiency and speed in metabolic engineering tasks. To try teemi, visit our Google Colab notebooks.
teemi not only simplifies the strain construction process but also offers the flexibility to adapt to different experimental workflows through its open-source Python platform. This allows for efficient automation of repetitive tasks and a faster pace in metabolic engineering.
Our demonstration of teemi in a complex machine learning-guided metabolic engineering task showcases its efficiency and speed by debottlenecking a crucial step in the strictosidine pathway. This highlights the versatility and usefulness of this tool in various biological applications.
Curious about how you can build strains easier and faster with teemi? Head over to our Google Colab notebooks and give it a try.
For a quick introduction, check our quick guides:
- A Quick Guide to Creating a Combinatorial Library
- A Quick Guide to making a CRISPR plasmid with USER cloning (for the beginner)
teemi has been published in PLOS COMPUTATIONAL BIOLOGY: "teemi: An open-source literate programming approach for iterative design-build-test-learn cycles in bioengineering". Please cite it if you've used teemi in a scientific publication.

- Features
- Getting started
- A Quick Guide to Creating a Combinatorial Library
- A Quick Guide to making a CRISPR plasmid with USER cloning (for the beginner)
- Colab notebooks
- Strictosidine case : First DBTL cycle
- Strictosidine case : Second DBTL cycle
- Installation
- Documentation and Examples
- Contributions
- License
- Credits
- Combinatorial library generation
- HT cloning and transformation workflows
- Flowbot One instructions
- CSV-based LIMS system as well as integration to Benchling
- Genotyping of microbial strains
- Advanced Machine Learning of biological datasets with the AutoML H2O
- Workflows for selecting enzyme homologs
- Promoter selection workflows from RNA-seq datasets
- Data analysis of large LC-MS datasets along with workflows for analysis
To get started with making microbial strains in an HT manner please follow the steps below:
- Install teemi. You will find the necessary information below for installation.
- Check out our notebooks for inspiration to make HT strain construction with teemi.
- You can start making your own workflows by importing teemi into either Google colab or Jupyter lab/notebooks.
This guide provides a simple example of the power and ease of use of the teemi tool. Let's take the example of creating a basic combinatorial library with the following design considerations:
- Four promoters
- Ten enzyme homologs
- A Kozak sequence integrated into the primers
Our goal is to assemble a library of promoters and enzymes into a genome via in vivo assembly. We already have a CRISPR plasmid; all we need to do is amplify the promoters and enzymes for the transformation. This requires generating primers and making PCRs. We'll use teemi for this process.
To begin, we load the genetic parts using Teemi's easy-to-use function read_genbank_files(), specifying the path to the genetic parts.
from teemi.design.fetch_sequences import read_genbank_files
path = '../data/genetic_parts/G8H_CYP_CPR_PARTS/'
pCPR_sites = read_genbank_files(path+'CPR_promoters.gb')
CPR_sites = read_genbank_files(path+'CPR_tCYC1.gb')We have four promoters and ten CPR homologs (all with integrated terminators).
We want to convert them into pydna.Dseqrecord objects from their current form as Bio.Seqrecord. We can do it this way:
from pydna.dseqrecord import Dseqrecord
pCPR_sites = [Dseqrecord(seq) for seq in pCPR_sites]
CPR_sites = [Dseqrecord(seq) for seq in CPR_sites]Next, we add these genetic parts to a list in the configuration we desire, with the promoters upstream of the enzyme homologs.
list_of_seqs = [pCPR_sites, CPR_sites]If we want to integrate a sgRNA site into the primers, we can do that. In this case, we want to integrate a Kozak sequence. We can initialize it as shown below.
kozak = [Dseqrecord('TCGGTC')]Now we're ready to create a combinatorial library of our 4x10 combinations. We can import the Teemi class for this.
from teemi.design.combinatorial_design import DesignAssemblyWe initialize with the sequences, the pad (where we want the pad - in this case, between the promoters and CPRs), then select the overlap and the desired temperature for the primers. Note that you can use your own primer calculator. Teemi has a function that can calculate primer Tm using NEB, for example, but for simplicity, we'll use the default calculator here.
CPR_combinatorial_library = DesignAssembly(list_of_seqs, pad = kozak , position_of_pads =[1], overlap=35, target_tm = 55 )Now, we can retrieve the library.
CPR_combinatorial_library.primer_list_to_dataframe()| id | anneals to | sequence | annealing temperature | length | price(DKK) | description | footprint | len_footprint |
|---|---|---|---|---|---|---|---|---|
| P001 | pMLS1 | ... | 56.11 | 20 | 36.0 | Anneals to pMLS1 | ... | 20 |
| P002 | pMLS1 | ... | 56.18 | 49 | 88.2 | Anneals to pMLS1, overlaps to 2349bp_PCR_prod | ... | 28 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
The result of this operation is a pandas DataFrame which will look similar to the given example (note that the actual DataFrame have more rows).
To obtain a DataFrame detailing the steps required for each PCR, we can use the following:
CPR_combinatorial_library.pcr_list_to_dataframe()| pcr_number | template | forward_primer | reverse_primer | f_tm | r_tm |
|---|---|---|---|---|---|
| PCR1 | pMLS1 | P001 | P002 | 56.11 | 56.18 |
| PCR2 | AhuCPR_tCYC1 | P003 | P004 | 53.04 | 53.50 |
| PCR3 | pMLS1 | P001 | P005 | 56.11 | 56.18 |
| ... | ... | ... | ... | ... | ... |
The output is a pandas DataFrame. This is a simplified version and the actual DataFrame can have more rows.
Teemi has many more functionalities. For instance, we can easily view the different combinations in our library.
CPR_combinatorial_library.show_variants_lib_df()| 0 | 1 | Systematic_name | Variant |
|---|---|---|---|
| pMLS1 | AhuCPR_tCYC1 | (1, 1) | 0 |
| pMLS1 | AanCPR_tCYC1 | (1, 2) | 1 |
| pMLS1 | CloCPR_tCYC1 | (1, 3) | 2 |
| ... | ... | ... | ... |
This command results in a pandas DataFrame, showing the combinations in the library. This is a simplified version and the actual DataFrame would have 40 rows for this example.
The next step is to head to the lab and build some strains. Luckily, we have many examples demonstrating how to do this for a large number of strains and a bigger library (1280 combinations). Please refer to our Colab notebooks below where we look at optimizing strictosidine production in yeast with Teemi.
Here is a quick guide on how we simulate the assembly of a CRISPR plasmid with USER cloning. Big thanks to Björn Johansson for the initial work with pydna that makes much of this possible. Please check out pydna here.
Let's begin with the simple workflow:
from pydna.primer import Primer
from pydna.dseqrecord import DseqrecordStep 1: Getting the fragments we want to integrate into our CRISPR plasmid. Specifically, we aim to integrate sgRNAs to knock out two targets.
# 1.1: Define the primers
U_pSNR52_Fw_1 = Primer('CGTGCGAUTCTTTGAAAAGATAATGTATGA')
TJOS_66_P2R = Primer('ACCTGCACUTAACTAATTACATGACTCGA')
U_pSNR52_Fw_2 = Primer('AGTGCAGGUTCTTTGAAAAGATAATGTATGA')
TJOS_65_P1R = Primer('CACGCGAUTAACTAATTACATGACTCGA')Primers are short, single-stranded DNA sequences that are necessary for targeting the specific DNA region we want to amplify using PCR.
1.2: Get the gRNA template. We retrieve the gRNA template from plate we have in the lab with the following teemi function. The gRNA template is the DNA sequence that encodes the guide RNA. This RNA molecule guides the Cas9 protein to the target DNA sequence, where it induces a cut.
from teemi.lims.csv_database import get_dna_from_box_name
gRNA1_template = get_dna_from_plate_name('gRNA1_template (1).fasta', 'plasmid_plates', database_path="G8H_CPR_library/data/06-lims/csv_database/")1.3: Perform a PCR to amplify the gRNA. PCR (Polymerase Chain Reaction) is a technique used to amplify a specific DNA sequence. Here, we're amplifying our gRNA templates.
from pydna.amplify import pcr
gRNA1_pcr_prod = pcr(U_pSNR52_Fw_1,TJOS_66_P2R, gRNA1_template)
gRNA2_pcr_prod = pcr(U_pSNR52_Fw_2,TJOS_65_P1R, gRNA2_template)1.4: Use the USER enzyme to process the PCR products. The USER enzyme is used to create single-stranded overhangs on the PCR products, which will facilitate their insertion into the plasmid.
from teemi.design.cloning import USER_enzyme
gRNA1_pcr_USER = USER_enzyme(gRNA1_pcr_prod)
gRNA2_pcr_USER = USER_enzyme(gRNA2_pcr_prod)
print(gRNA1_pcr_USER)
print(gRNA2_pcr_USER)Output:
Dseq(-425)
TCTT..GTTAAGTGCAGGT
GCACGCTAAGAA..CAAT
Dseq(-425)
TCTT..GTTAATCGCGTG
TCACGTCCAAGAA..CAAT
Step 2: Digesting the plasmid. The plasmid is a small, circular DNA molecule. We're importing a specific template that we'll use to integrate our gRNAs.
# 2.1: Import the plasmid
vector = Dseqrecord(get_dna_from_plate_name('Backbone_template - p0056_(pESC-LEU-ccdB-USER) (1).fasta', 'plasmid_plates', database_path="G8H_CPR_library/data/06-lims/csv_database/"), circular = True)2.2: Digest the plasmid with AsiSI enzyme. Digestion with the AsiSI enzyme creates specific cuts in the plasmid, allowing us to insert our gRNAs at these locations.
from Bio.Restriction import AsiSI
vector_asiSI, cCCDB = sorted( vector.cut(AsiSI), reverse=True)
print(vector_asiSI.seq)Output:
Dseq(-6972) CGCG..TGCGAT TAGCGC..ACGC
2.3: Nick the digested plasmid using a nicking enzyme
from teemi.design.cloning import nicking_enzyme
vector_asiSI_nick = Dseqrecord(nicking_enzyme(vector_asiSI))
vector_asiSI_nick.seqNicking enzymes create single-stranded breaks in the DNA. This step prepares the plasmid for the insertion of the gRNAs.
Output:
Dseq(-6972)
CATT..AATGCGTGCGAT
TAGCGCACGTAA..TTAC
Step 3: Assembling sgRNAs and vector
# 3.1: Combine the nicked vector with the USER processed gRNAs and loop the resulting sequence
rec_vec = (vector_asiSI_nick + gRNA1_pcr_USER + gRNA2_pcr_USER).looped()
rec_vec.seqIn this final step, we're assembling the plasmid by combining the nicked vector with the processed gRNAs. The resulting molecule is a circular DNA plasmid containing our gRNAs.
Output:
Dseq(o7797) CATT..CGTG GTAA..GCAC
For more real-life examples on how to use this in complex metabolic worklfows in a high-throughput manner pleas check our Colab notebooks .
As a proof of concept we wanted to show how teemi and literate programming can be used to streamline bioengineering workflows. These workflows should serve as a guide or a help to build your own.
Specifically, in this first study we present how we used teemi and literate programming to build simulation-guided, iterative, laboratory workflows for optimizing strictosidine production in yeast. If you wanna read the study you can find the pre-print here.
Below you can find all the notebooks developed in this work. Just click the Google colab badge to start the workflows.
The strictosidine pathway and short intro: Strictosidine is a crucial precursor for 3,000+ bioactive alkaloids found in plants, used in medical treatments like cancer and malaria. Chemically synthesizing or extracting them is challenging. We're exploring biotechnological methods to produce them in yeast cell factories. But complex P450-mediated hydroxylations limit production. We're optimizing these reactions using combinatorial optimization, starting with geraniol hydroxylation(G8H) as a test case. Feal free to check out the notebooks for more information on how we did it.
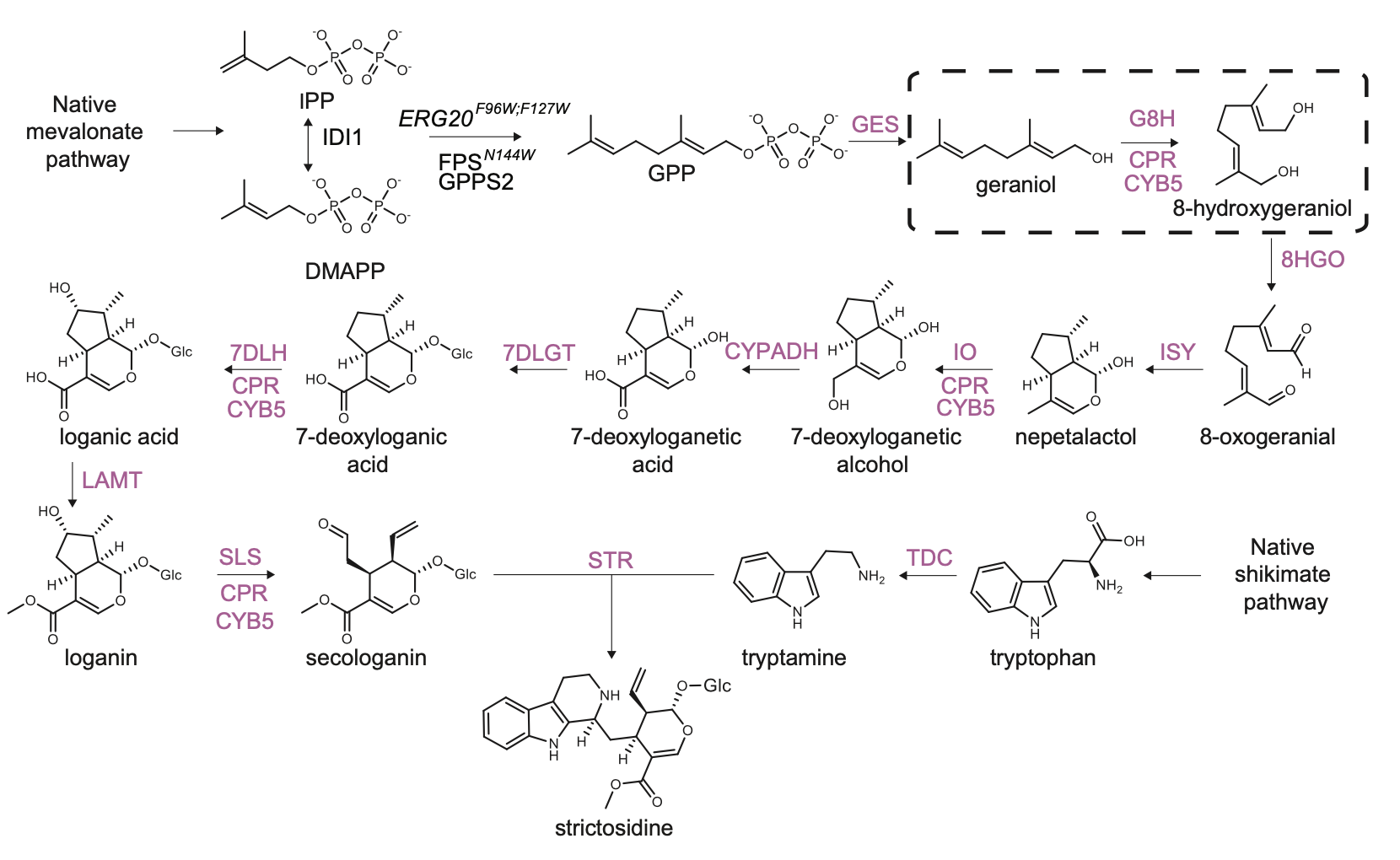
DESIGN:
- Automatically fetch homologs from NCBI from a query in a standardizable and repeatable way
- Promoters can be selected from RNAseq data and fetched from online database with various quality measurements implemented
- Combinatorial libraries can be generated with the DesignAssembly class along with robot executable intructions
BUILD:
- Assembly of a CRISPR plasmid with USER cloning
- Construction of the background strain by K/O of G8H and CPR
- First combinatorial library was generated for 1280 possible combinations
TEST:
- Data processing of LC-MS data and genotyping of the generated strains
LEARN:
- Use AutoML to predict the best combinations for a targeted second round of library construction
DESIGN:
- Results from the ML can be translated into making a targeted library of strains
BUILD:
- Shows the construction of a targeted library of strains
TEST:
- Data processing of LC-MS data like in notebook 6
LEARN:
- Second ML cycle of ML showing how the model increased performance and saturation of best performing strains
Use pip to install teemi from PyPI.
$ pip install teemi
If you want to develop or if you cloned the repository from our GitHub you can install teemi in the following way.
$ pip install -e <path-to-teemi-repo>
Or if you are in the teemi repository:
$ pip install -e .
For those who want to contribute or develop further, you can install the development version with:
$ pip install -e .[dev]
Or directly from PyPI:
$ pip install teemi[dev]
You might need to run these commands with administrative
privileges if you're not using a virtual environment (using sudo for example).
Please check the documentation
for further details.
Documentation is available on through numerous Google Colab notebooks with examples on how to use teemi and how we use these notebooks for strain construnction. The Colab notebooks can be found here teemi.notebooks.
- Documentation: https://teemi.readthedocs.io
Contributions are very welcome! Check our guidelines for instructions how to contribute.
- Free software: MIT license
- This package was created with Cookiecutter and the audreyr/cookiecutter-pypackage project template.
- teemis logo was made by Jonas Krogh Fischer. Check out his website.