- Simulated Data
- Min Hash algorithm implemented in C++
- Containment Hash algorithm implemented in C++
- Mapper function
- Generate Simulated Data:
generate_data.py - Min Hash and Containment Hash algorithms:
hash.cpp, hash.h - Comparing both approaches:
main.cpp - Plotting graphs:
draw_graphs.py - Mapper function:
mapper.cpp - Scripts:
run.sh, query.sh
- Taxonomic classification
- To detect presence or absence of genome in metagenomics sample
- To detect the presence of very small, low abundance microorganisms in a metagenomic data set
- Estimate Jaccard Index by running Min Hash and Containment Min Hash.
./run.sh <file1> <file2> <kmer_size> <hash_functions>
<false_positive_rate> <number_appended_to_results_files> <order_of_len_A> <order_of_len_B>./run.sh data/file3.txt data/file4.txt 20 200 0.01 2 150 1000
./run.sh data/file1.txt data/file2.txt 18 1000 0.02 3 15 1000Sample output:
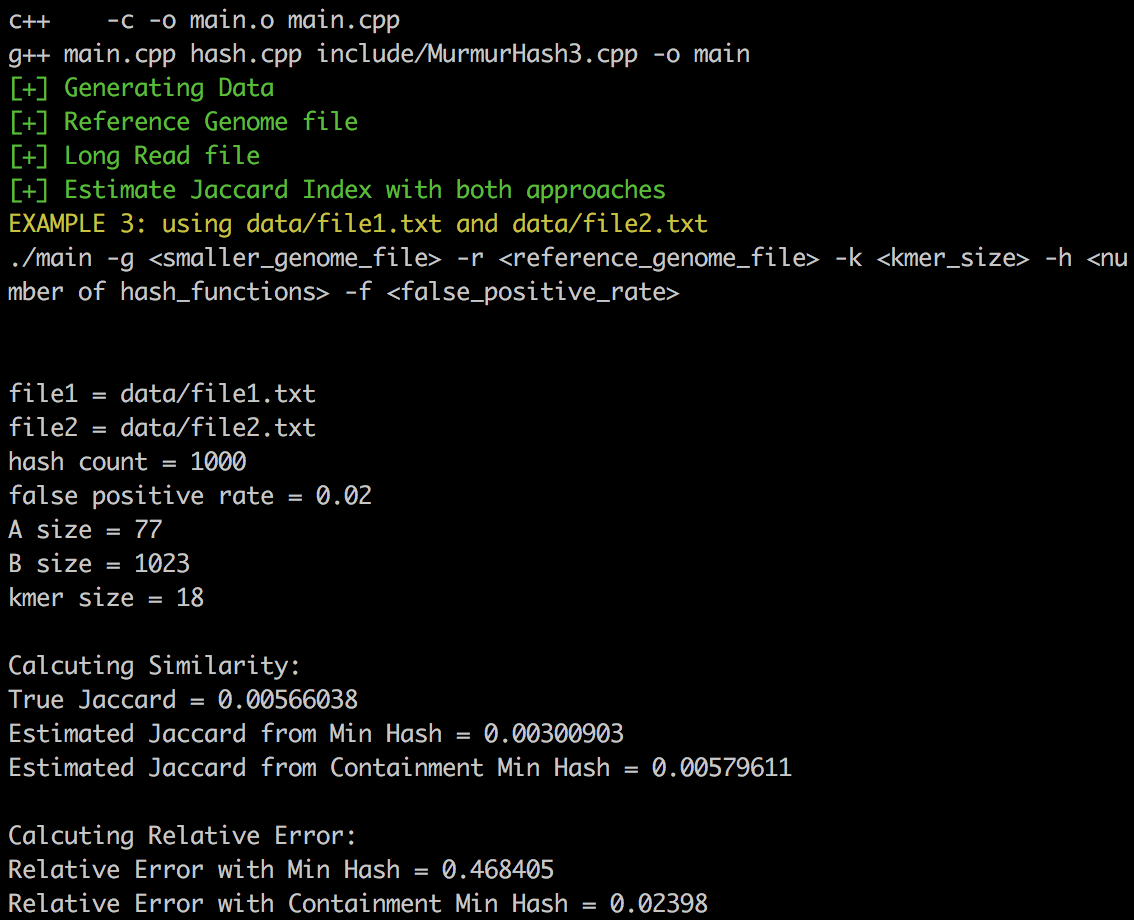
- Compare both approaches. Reproduce the graphs in the report.
./run.sh data/filex3.txt data/filex4.txt 20 150 0.04 3
./run.sh data/filex1.txt data/filex2.txt 25 1000 0.01 4Sample output:
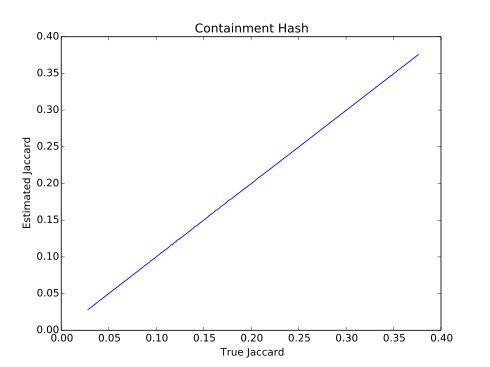
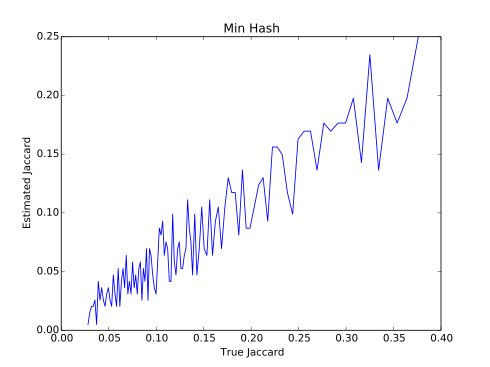
- Mapping long reads: Script generates reference genome and long reads and then maps long reads to reference genome if they have similarity higher than threshold.
./query.sh <long_reads_file> <reference_genome_file> <threshold>
<kmer_size> <hash_functions> <false_positive_rate> <number_of_long_reads_to_generate>./query.sh data/reference.txt data/longreads.txt 0.05 18 100 0.01 5
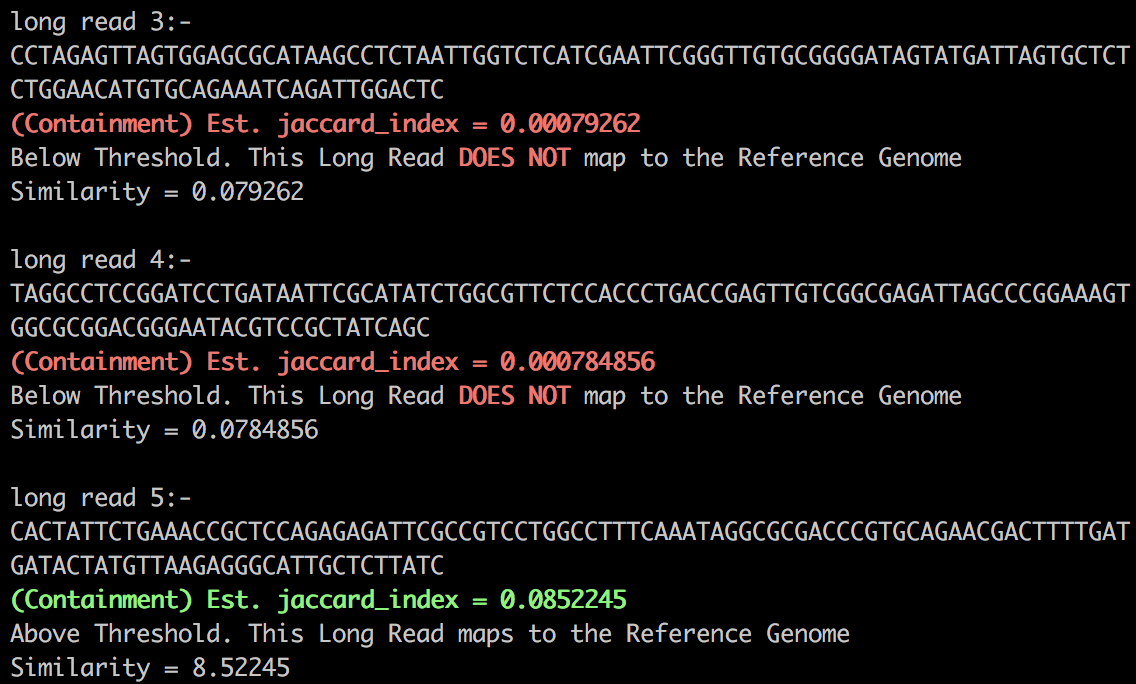
./query.sh data/reference.txt data/longreads.txt 0.05 20 200 0.02 10Sample output:
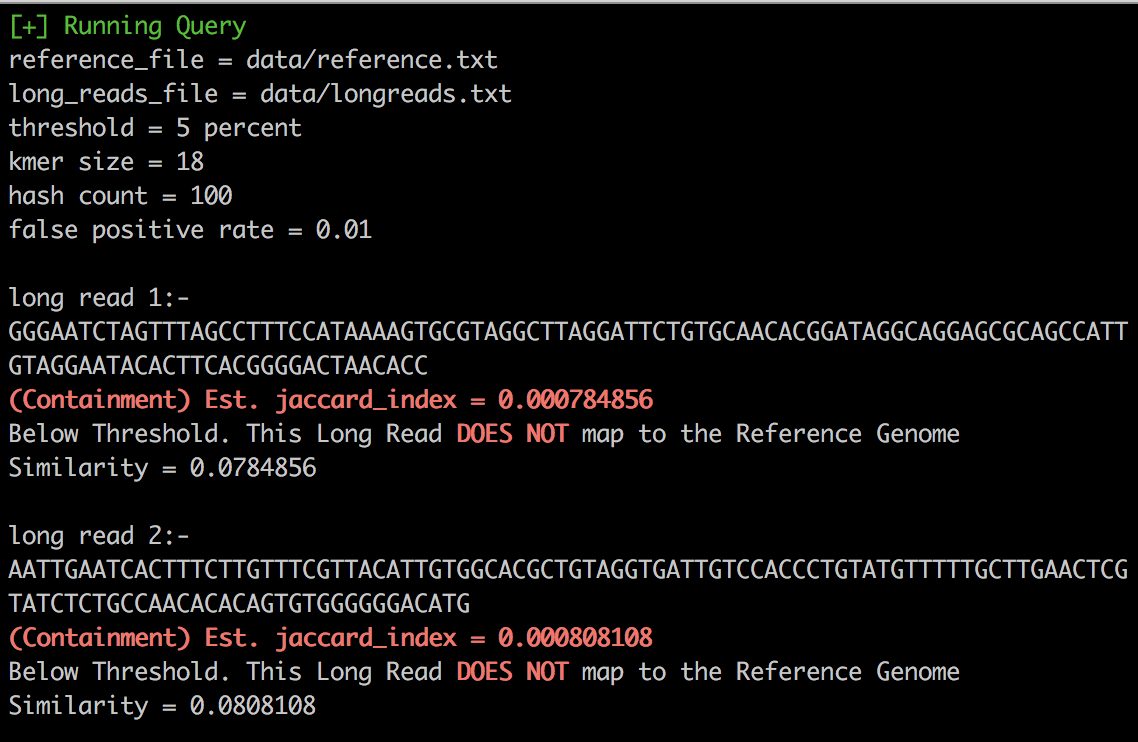
- Paper on Containment Min Hash approach: https://www.biorxiv.org/content/biorxiv/early/2017/09/04/184150.full.pdf
- Paper's github repo: https://github.com/dkoslicki/MinHashMetagenomics
- Mining Massive Data sets: http://infolab.stanford.edu/~ullman/mmds/ch3.pdf
- Hash Functions generation https://www.eecs.harvard.edu/~michaelm/postscripts/rsa2008.pdf
- Bloom filter http://www.partow.net/programming/bloomfilter/index.html
- Bloom filter https://archive.codeplex.com/?p=libbloom
- Murmur Hash3 https://github.com/aappleby/smhasher
- Mapping reads http://sfg.stanford.edu/mapping.html
- C++ notes: https://www.uow.edu.au/~lukes/TEXTBOOK/notes-cpp/