For inquiries, contact [email protected], [email protected], or [email protected]
This repo contains a preprocess.py and GASR.py for swift assembly of short read sequence datasets given a reference seed sequence.
GASR.py contains an option NO_GAP that allows a user to toggle between adding bases without an nmer gap or with one. Some short read datasets may require the option to be toggled on for the assembler to work properly.
Our python script relies on the BioPython toolkit suite to quickly grep large short read sequence files.
Before running any of the scripts, install the required dependencies from requirements.txt by running pip install -r requirements.txt or create a separate conda environment and run conda install --file requirements.txt.
usage: GASR.py [-h] [--input INPUT] [--seedseq SEEDSEQ] [--nmer NMER] [--maxround MAXROUND] [--outdir OUTDIR]
[--process_multiple_fastq PROCESS_MULTIPLE_FASTQ] [--no_gap NO_GAP]
options:
-h, --help show this help message and exit
--input INPUT input fastq sequence (preprocessed or not) to assemble
--seedseq SEEDSEQ seed sequence from where we will begin our search (e.g. GTGCTTCACCAACGTGTACGCCGACAGCTTCGTG)
--nmer NMER size of kmer to complete searching rounds (default = 25)
--maxround MAXROUND maximum number of searching rounds (default = 65)
--outdir OUTDIR path to directory to save output files (default is cwd)
--process_multiple_fastq PROCESS_MULTIPLE_FASTQ
True if input files need to be preprocessed. Must input multiple fastq files, unprocesed separated by a space (default = False)
--no_gap NO_GAP True if you want to extend without using any gaps (default = False)
python GASR.py --input 1_Mod500pgMS0pg_S1_L001_R1_001_final_combined.fastq --outdir example_output --seedseq GGTTCGACAACCCCGTGCTGCCCTTCAACGACGGCGTGTACTTC
python GASR.py --input '1_Mod500pgMS0pg_S1_L001_R1_001.fastq 1_Mod500pgMS0pg_S1_L001_R2_001.fastq' --outdir example_output --process_multiple_fastq True --seedseq GGTTCGACAACCCCGTGCTGCCCTTCAACGACGGCGTGTACTTC
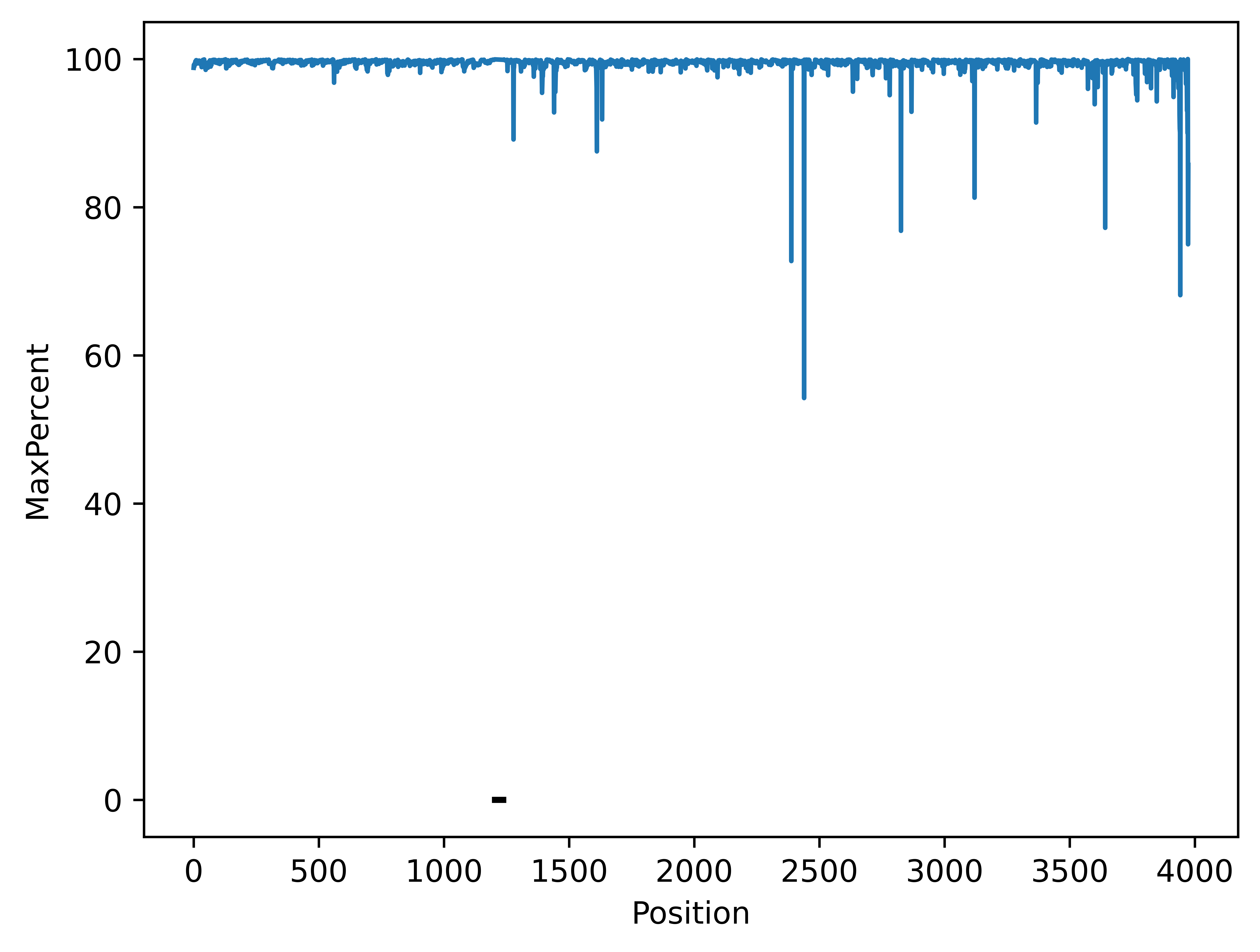
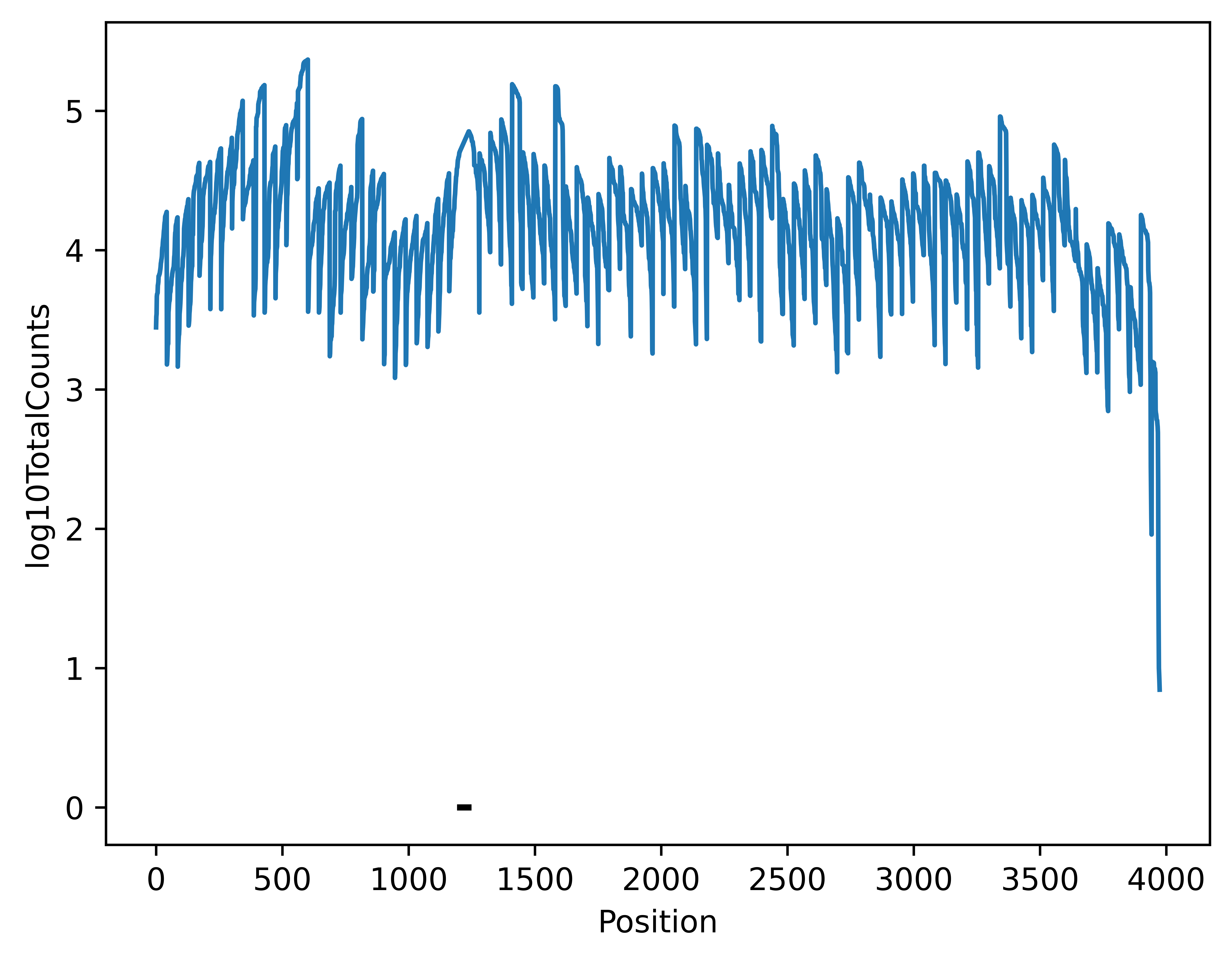
The script should take no more than 2-10 minutes to run. We have tested with large (~1 gb) input files and a variety of test input seed sequences. In the output folder specified (or cwd if not), we also output two figures and a Final_assembled_info.csv for scrutiny. Final_assembled_info.csv contains a detailed position-specific description for each base assembled. It contains a representative base column (i.e. the base with the highest consistency across reads matching the grepped sequences) as well as broken down counts and percentages of each positions possible base identity from the grepped searches.
The topmost figure displays a metric for confidence in sequence position up- and downstream of your input seed sequence (shown as the black bar). The bottommost figure displays the number of logFold counts of reads we see in the short read .fastq file for each sequence position up- and downstream of the seed sequence.